Research
Poster of our laboratory
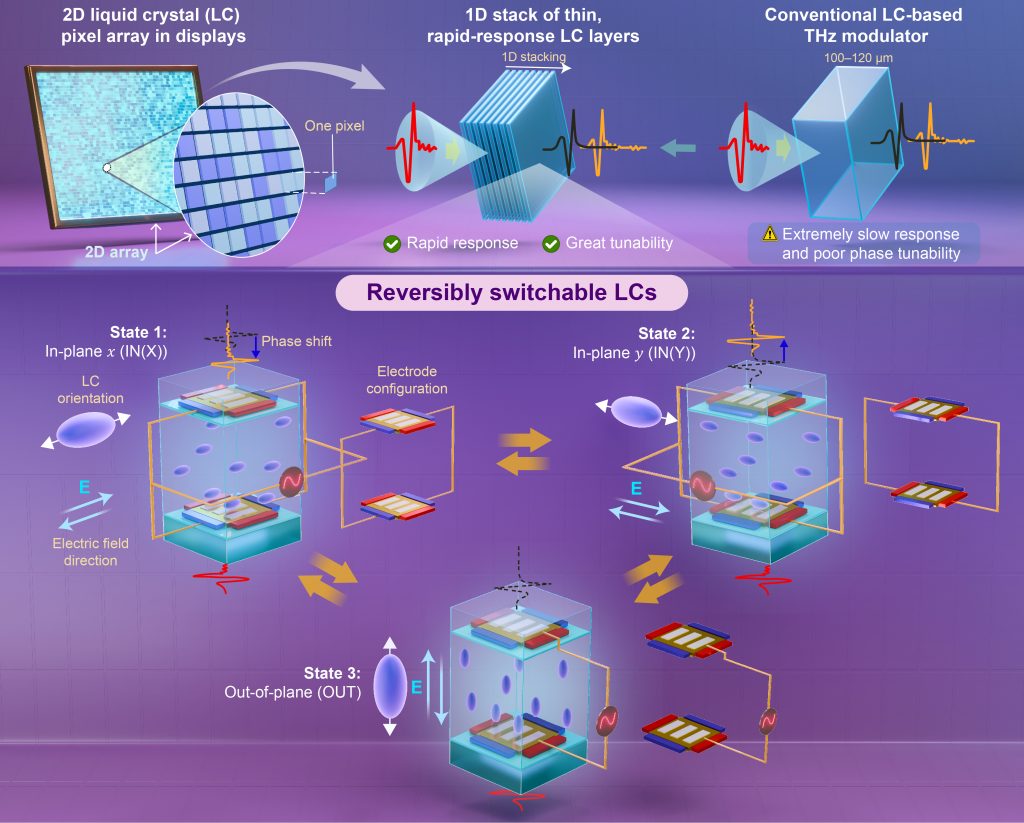
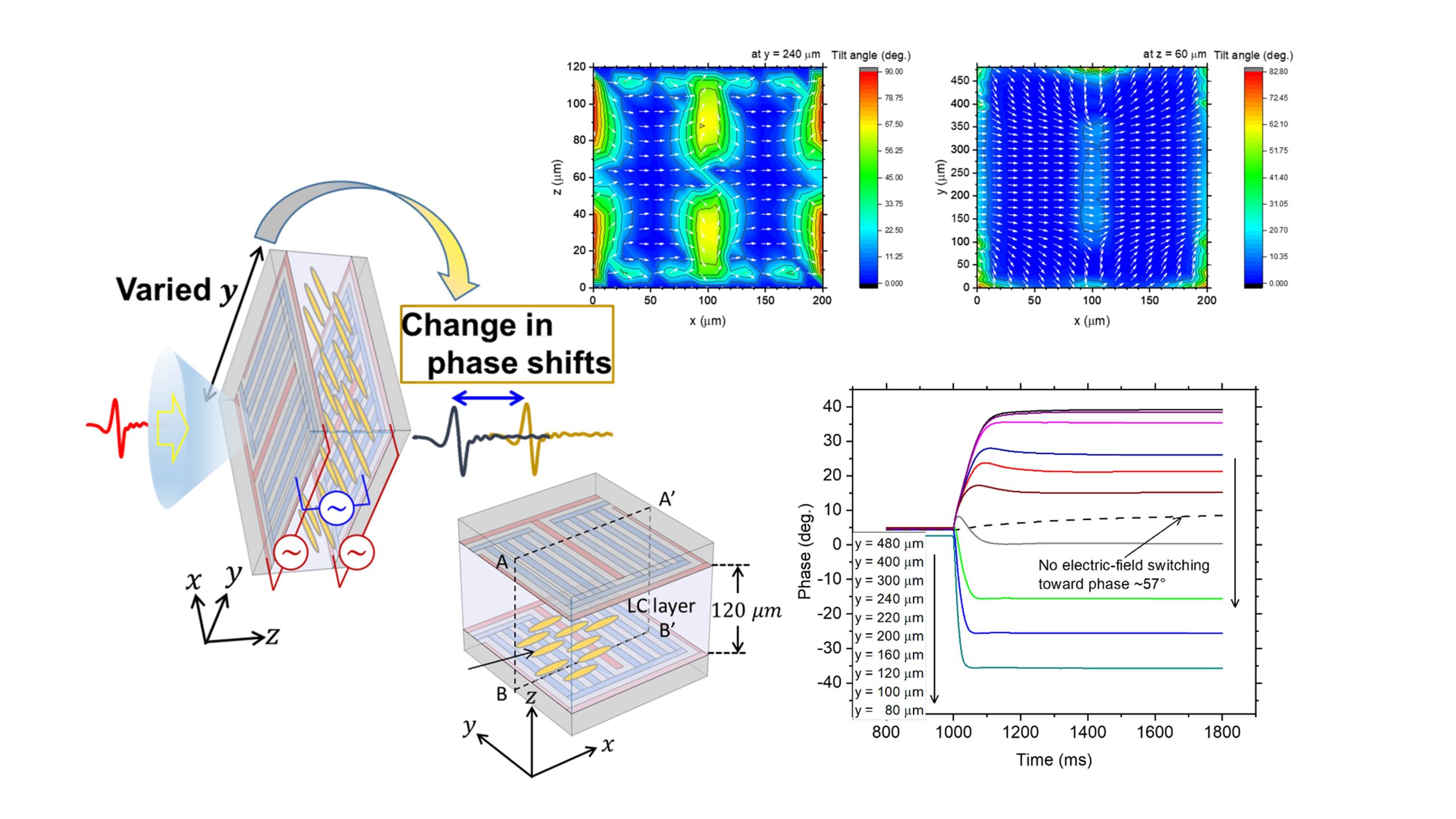
Optimizing the mechanism for modulating THz phases by reversibly switching layers of liquid crystals between two in-plane and one out-of-plane states
We previously proposed electrode structures to reversibly switch liquid crystals (LCs), which respond reasonably rapidly, between three orthogonal orientational states. Here, to leverage the excellent inherent tunability of LCs for use in terahertz (THz) phase modulators, we reframe the working principles of these electrode structures for driving a thick LC layer, and we investigate how low-voltage operation affects the switching behavior. Phase changes of 100° are demonstrated, but in principle, the phase range can be broadened by increasing the retardation of the LC medium. According to the operating principles, a pair of electrodes with identical layouts must precisely mirror each other on the inner surfaces of the top and bottom substrates separated by a gap; however, misaligning the two substrates scarcely affects the switching characteristics, providing wiggle room for sufficiently accurate alignment. Statically, even a low voltage enables switching one state to another while reaching the maximum phase shift. By contrast, the dynamic responses to low voltages degrade and are extremely slow. Further, although high-voltage operation provides reasonable response times between the three states, it is incompatible with continuous tunability. This problem prompted us to consider a stack of thin, rapid-response LC layers and to simultaneously or independently switch each layer to create a continuous or discrete range of possible phase shifts, respectively. In other words, replacing a two-dimensional pixel array, like a display, with a one-dimensional stack of LC layers would enable tunability compatible with reasonably fast responses, suggesting a technical advance toward realizing LC-based THz modulators.
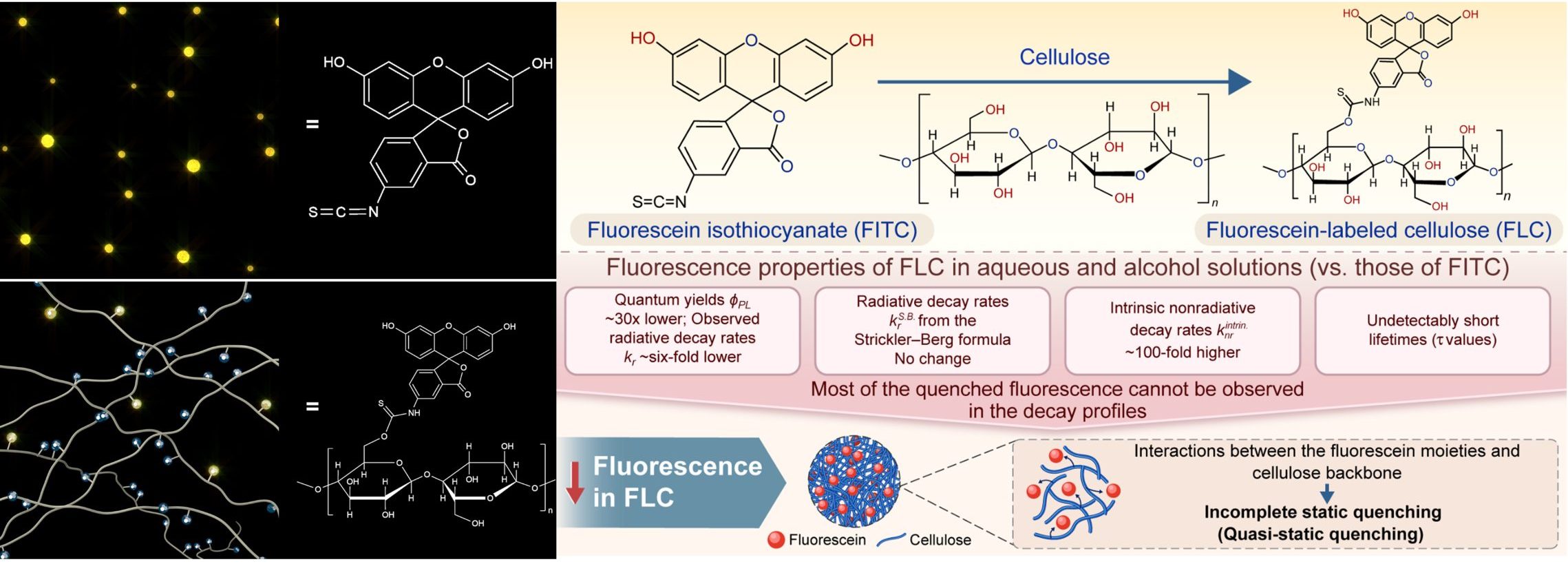
Characterizing the photoluminescence of fluorescein-labeled cellulose in aqueous and alcohol solutions: Influence of the cellulose backbone
Although many dyes have been introduced into cellulose, whether bound to its backbone or within a cellulose matrix, few studies have determined whether the backbone statically or dynamically quenches the photoluminescence of the dye. To advance cellulosic fluorescent films, the influence of the cellulose backbone on photoluminescence must be understood. We determined the fluorescence properties of fluorescein isothiocyanate (FITC) and fluorescein-labeled cellulose (FLC) in water and alcohol, including their quantum yields ɸPL, lifetimes τ, and rates of radiative kr and nonradiative knr decay. Dissolved FLC had a ~30× lower ɸPL than FITC, suggesting that incorporating FITC into the cellulose backbone remarkably reduces the fluorescence efficiency. The FLC solutions had a 6-fold lower kr than their FITC counterparts but a 10–20 times higher knr. Presumably, this was because the cellulose backbone interacted weakly with the fluorescein moieties, suggesting a quenching mechanism that can be termed quasi-static, corresponding to static quenching between the fluorescein moieties and cellulose backbone, in addition to the fluorescence quenching caused by the intramolecular nonradiative processes of fluorescein, as observed in conventional molecules. Using the Strickler‒Berg formula, we deduced the analytical radiative decay rate constants krS.B. and eventually estimated the number of very short-lived fluorescein moieties per single fluorescent fluorescein moiety, corresponding well with static quenching.
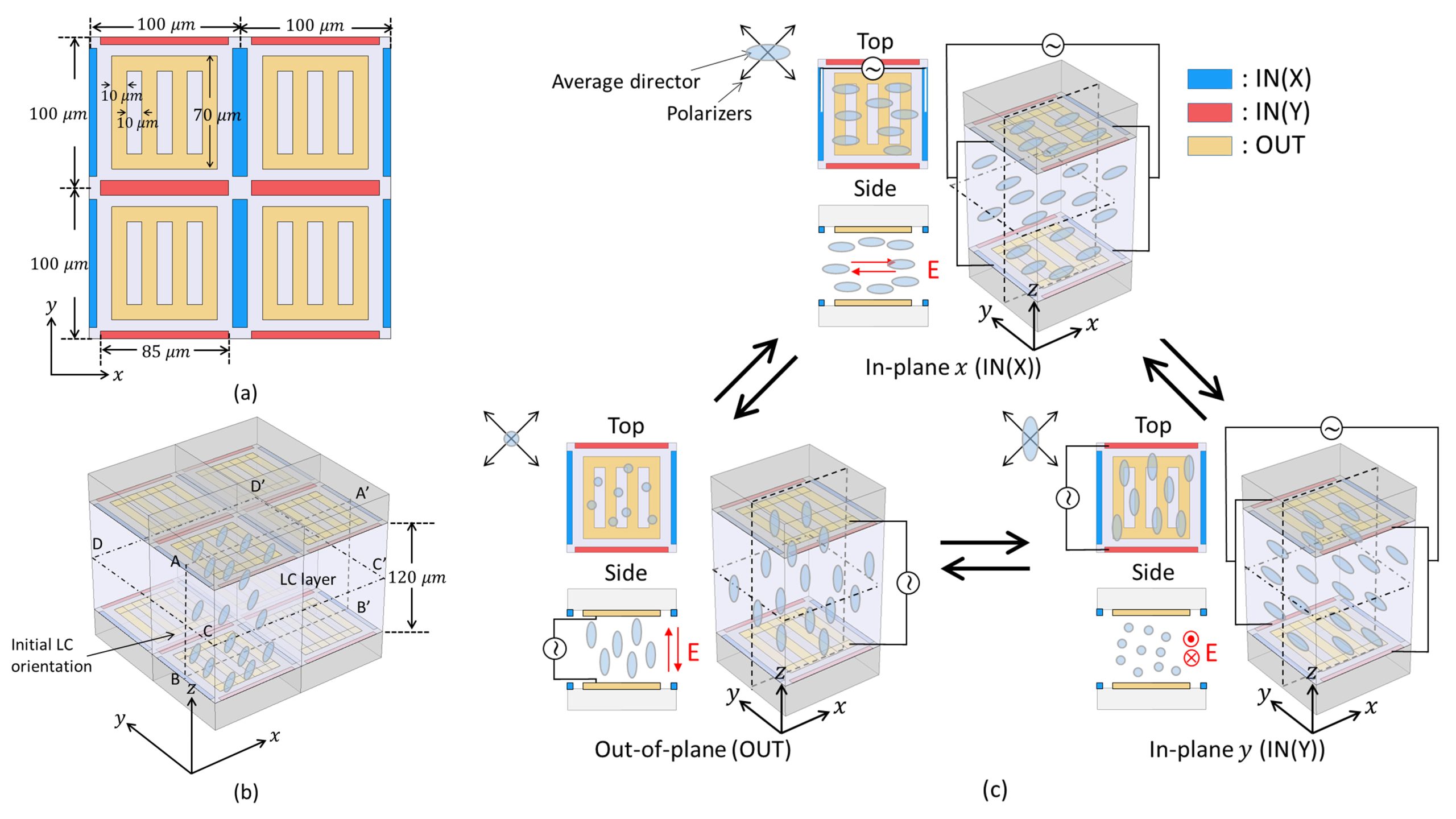
Reversibly switching liquid crystals between three orthogonal orientation states for use in rapid-response THz phase shifters
Liquid crystal (LC) devices for terahertz phase shifters inevitably use a thick cell gap for the required retardation, severely delaying the LC response. To improve the response, we virtually demonstrate novel LC switching between in-plane and out-of-plane for reversible switching between three orthogonal orientation states, broadening the range of continuous phase shifts. This LC switching is realized using a pair of substrates, each with two pairs of orthogonal finger-type electrodes and one grating-type electrode for in- and out-of-plane switching. An applied voltage generates an electric field that drives each switching process between the three distinct orientation states, enabling a rapid response.
Kinetic arrest during the drying of cellulose nanocrystal films from aqueous suspensions analogous to the freezing of thermal motions (Cellulose photonics):
This study aims to resolve some gaps in the knowledge on structural color in cellulose nanocrystal (CNC) films by drawing on our own observations of liquid crystal (LC) formation in CNC suspensions and previous studies on the formation of such films. We believe that our study makes a significant contribution to the literature because we systematically study the formation of these films and the self-organization of CNCs during evaporation under various conditions. We combine our understanding of pitch and ordering in CNC LCs in suspension with an analogy to the kinetic processes at play in thermotropic LCs to advance our understanding of the mechanism underlying this coloration, which is the subject of ongoing debate. Interestingly, we find that the solvent (water in our case) plays a role analogous to that of temperature in thermotropic LCs, e.g., those formed in aqueous CNC suspensions.
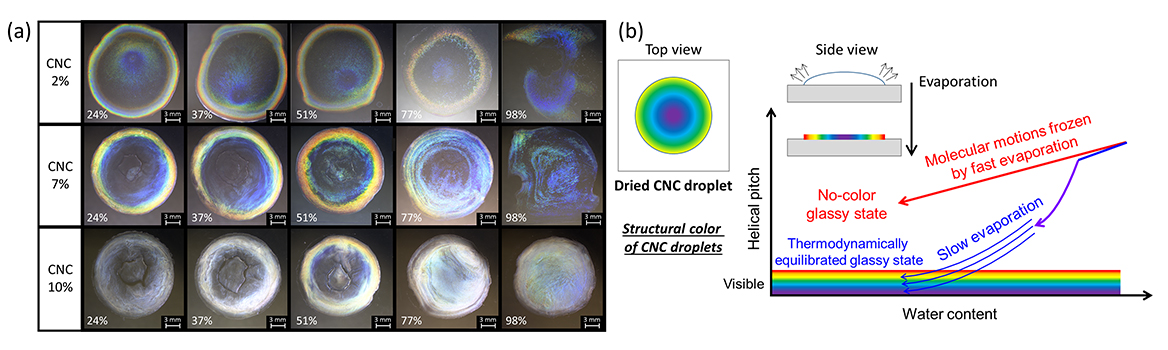
(b) Qualitative model of the effects of fast and slow water evaporation on the helical pitch.
Discovering and inventing innovative LC switching for THz electromagnetic waves
To unveil a novel switching mechanism in LC-based phase shifters for the THz range, we analyze how varying the dimensions of the electrode structures for THz in-plane and THz out-plane (TIP-TOP) switching influences the LC in-plane states with the corresponding phase shifts by calculating these effects in virtual devices. We have discovered that the most extreme dimensional effect produces a novel type of LC switching, which results in hexadirectional switching between the initial, intrinsic in-plane, and out-of-plane reorientations of the LCs, suggesting a broader range of phase shifts while maintaining a rapid response. This innovative finding may be exploited as a new type of LC switching for THz waves. We filed a patent in the US, Taiwan, and China and are currently filing applications in Japan as well. We have received a Notice of Allowance from USPTO on Nov. 2, 2022.
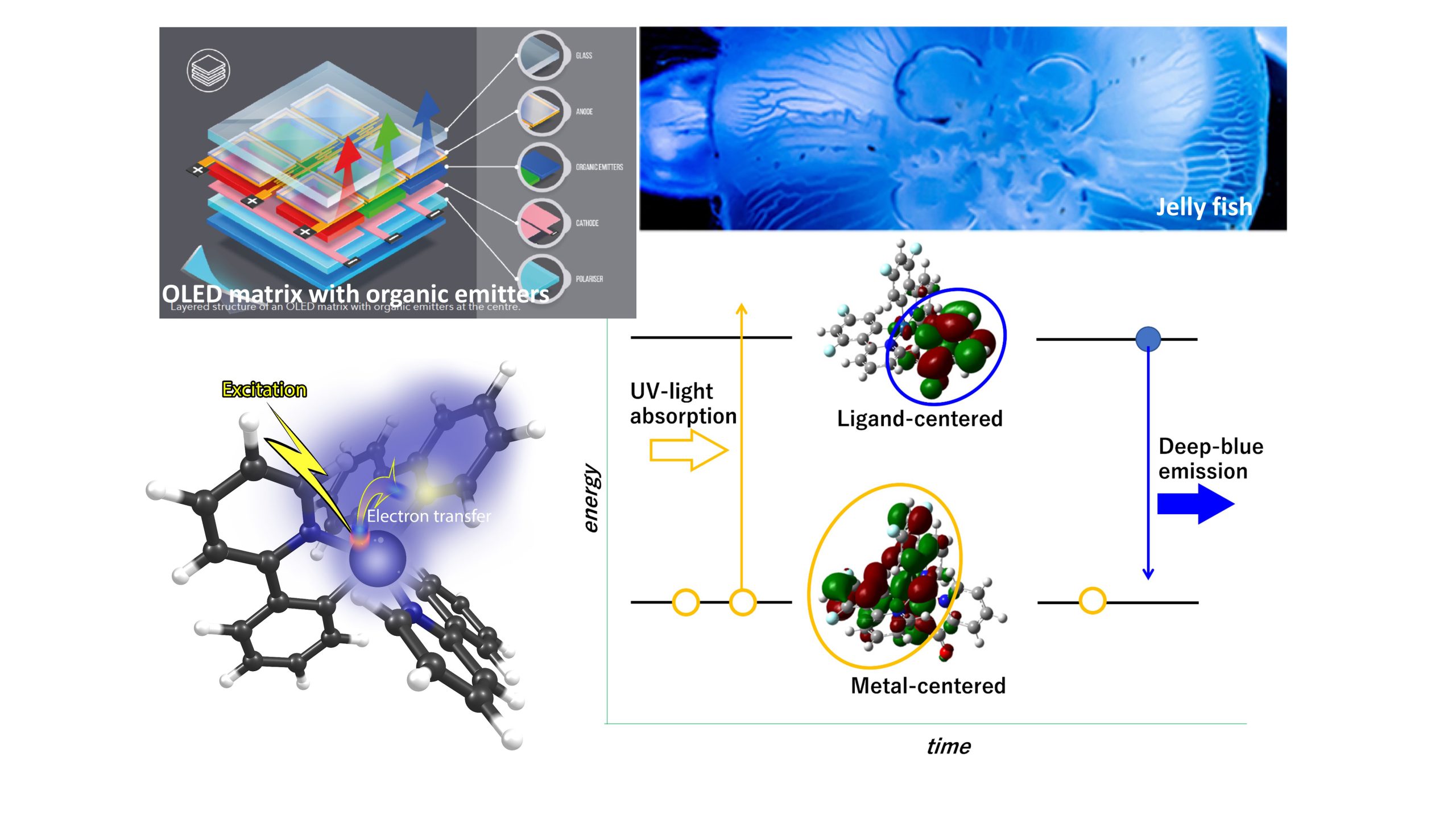
Unravelling the mysteries of deep-blue luminescence
To advance the a posteriori quest for deep-blue phosphorescence, we quantify the degree of metal-to-ligand charge transfer (MLCT) upon excitation using density functional theory calculations. Through quantifying the degree of MLCT as a guiding indicator, our manuscript essentially proposes a model that explains why deep-blue phosphorescence has not yet been attained and what the difficulties are in reaching this target. Our unique approach and the model underlying the study may attract the interest of a wide scientific community. A reviewer positively evaluated the potential of this article as an important contribution.
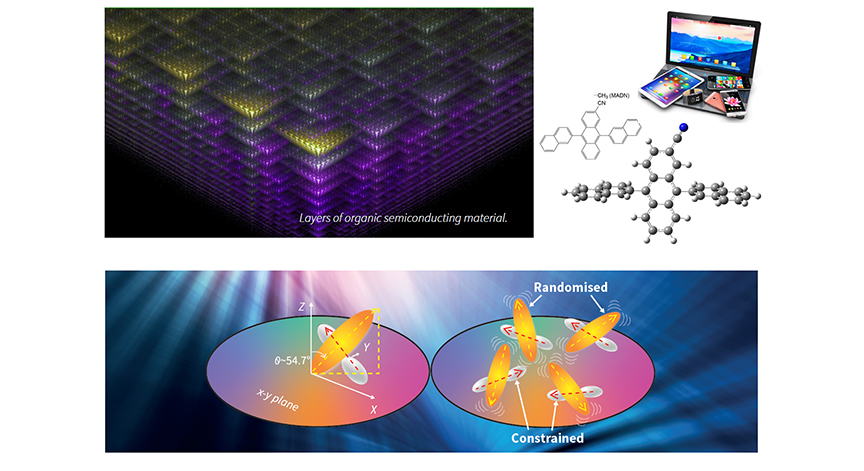
Intriguing molecular discoveries in display materials
Within a thin film of 2-cyano-9,10-di(2-naphthyl)anthracene (CADN), we have found that as the temperature increases and approaches the material’s phase-transition temperature, the motion of one part of the molecule increases, while another part becomes more ordered, which was reported for the first time worldwide. Variable-angle spectroscopic ellipsometry and second-harmonic generation were used to study changes in the molecular alignment in vapor-deposited organic thin films as samples were cyclically heated and cooled between room temperature (RT) and the phase-transition temperature.
with increasing temperature (right).
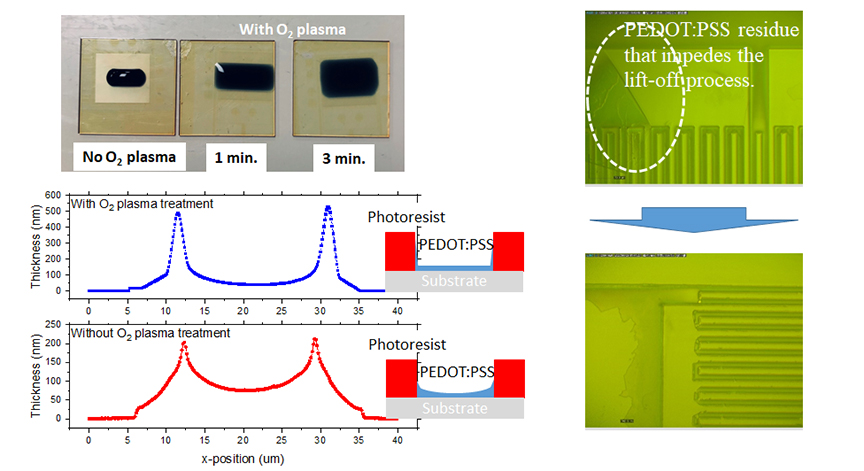
Effects of O2 plasma treatments on the photolithographic patterning of PEDOT:PSS
Poly (3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) is known for its potential to replace indium–tin oxide in various devices. Herein, when fabricating finger-type PEDOT:PSS electrodes using conventional photolithography, the cross-sectional profiles of the patterns are U-shaped instead of rectangular. The films initially suffer from non-uniformity and fragility as well as defects owing to undesirable patterns. Adding a small amount of hydrolyzed silane crosslinker to PEDOT:PSS suspensions increases the mechanical durability of PEDOT:PSS patterns while lifting off the photoresist. To further improve their microfabrication, we observe the effects of two additional oxygen (O2) plasma treatments on conventional photolithography processes for patterning PEDOT:PSS, expecting to observe how O2 plasma increases the uniformity of the patterns and changes the thickness and U-shaped cross-sectional profiles of the patterns. Appropriately exposing the patterned photoresist to O2 plasma before spin-coating PEDOT:PSS improves the wettability of its surface, including its sidewalls, and a similar treatment before lifting off the photoresist helps partially remove the spin-coated PEDOT:PSS that impedes the lift-off process. These two additional processes enable fabricating more uniform, defect-free PEDOT:PSS patterns. Both increasing the wettability of the photoresist patters before spin-coating PEDOT:PSS and reducing its conformal coverage are key to improving the photolithographic microfabrication of PEDOT:PSS.
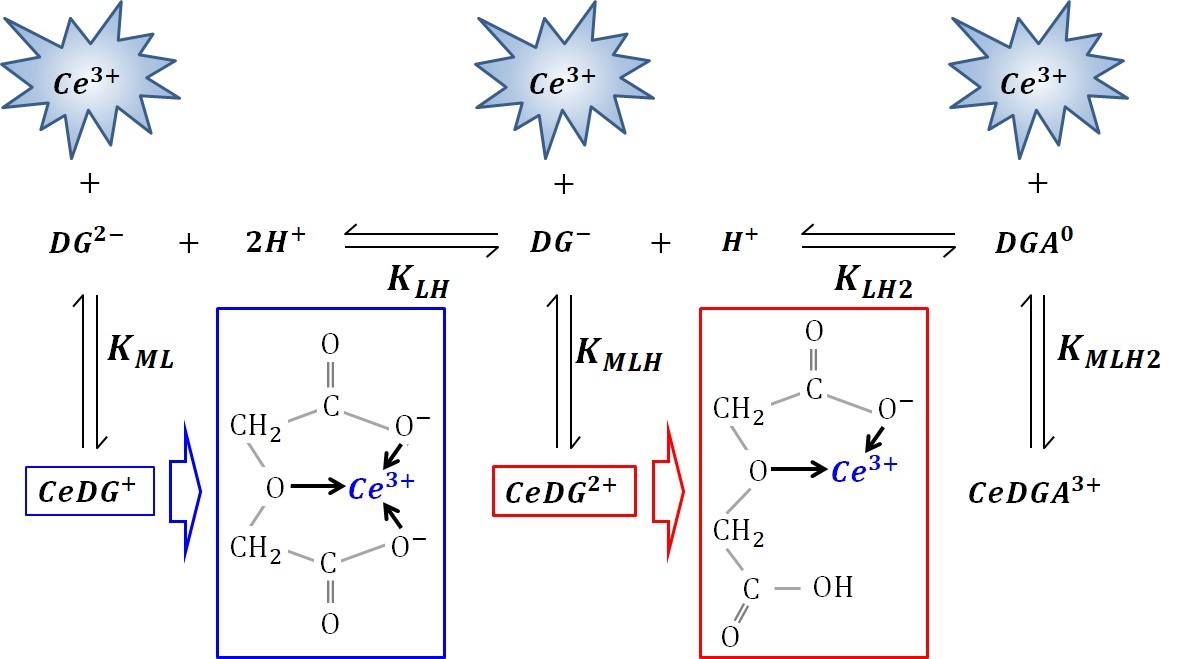
Interactions between hydrated cerium(III) cations and carboxylates in aqueous solution: Anomalously strong complex formation with diglycolate suggesting a chelate effect
Interactions between hydrated Ce3+ and various carboxylates are of fundamental interest. Anomalously strong interactions with Ce3+ occur when adding diglycolic acid (DGA) into a Ce3+ aqueous solution, unlike various other carboxylic acids. The complex formation constants of Ce3+ with these acids are evaluated via absorption and emission spectra. Hydrated Ce3+ emits fluorescence with unity quantum yield; however, adding various carboxylates statically quenches the fluorescence when Ce3+‒carboxylate complexes form, because the fluorescence lifetime is constant irrespective of the carboxylate concentration. In the observed static quenching, the complex formation constants obtained from the absorption and emission spectra (Kabs and Kem) agree well. The binding Ce3+ by the conjugate Lewis bases, i.e., carboxylates is approximately inversely proportional to the pH. Adding DGA into the system also statically quenches the fluorescence, but far more efficiently, even in a much weaker solution. We rigorously deduce Kabs and Kem of Ce3+ with DGA without any approximation using comparable concentrations. Careful fittings provide equivalent Kem and Kabs values, and varying the pH and ionic strength confirms this equivalence is an inherent property of the Ce3+-DGA system. Lewis acid–base theory cannot explain DGA binds Ce3+ ~1000 times more strongly than the other carboxylates. This anomalously strong binding may be due to a chelate effect caused by the DGA’s central oxygen atom, which forms a five-membered ring with the conjugate Lewis bases of DGA; double chelate rings can also form, while bis-deprotonated DGA binds Ce3+, facilitated by the central oxygen. Therefore, DGA enables efficient quenching through the chelate effect when it binds Ce3+.
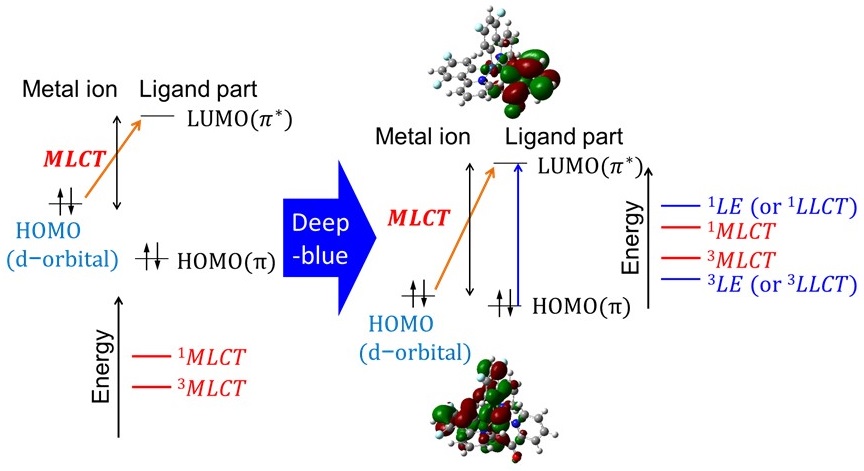
Advancing the a posteriori quest for deep-blue phosphorescence: Quantifying excitation-induced metal-to-ligand charge transfer as a guiding indicator
To advance the a posteriori quest for deep-blue phosphorescence, we quantify the degree of metal-to-ligand charge trans-fer (MLCT) upon excitation using density functional theory calculations. We also comprehensively determine how the MLCT nature of a state is correlated with the wavelength of potential phosphorescence through comparing the results of two typical ligand structures, i.e., 2-phenylpyridine and 1-phenyl-3-methylimidazolin-2-ylidene, with those of their derivatives. The MLCT nature can be defined as the difference in the electron density over the central metal ion that contributes to the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively). This study focuses on how the MLCT nature can be characterized upon excitation depending on the admixtures of molecular orbitals and how a stronger MLCT nature is preferable for efficient phosphorescence, while the MLCT nature exhibits a trade-off relationship with the wavelengths of phosphorescence in the blue region. Moreover, we elucidate how iridium(III) complexes are suitable for efficient phosphorescence and discuss a conceptual grand design for transition-metal complexes with deep-blue phosphorescence using the ligand field theory. We examine why strong MLCT nature upon excitation does not necessarily ensure highly efficient phosphorescence, taking into account possible relative energy gaps between the singlet MLCT, triplet locally excited, and d‒d transition states in a transition-metal complex formed after excitation. Nevertheless, the initial MLCT serves as a necessary condition that induces corollary energy-state configurations as well as decay from the excited states. Therefore, quantifying the nature of that MLCT, which is the first process upon excitation, with the possible wavelength of phosphorescence, is a useful parameter for a conceptual grand design for candidate complexes that can efficiently emit deep-blue phosphorescence. This reverse method of analyzing candidate complexes before experimentally formulating and studying them can accelerate the a posteriori quest for deep-blue-emitting materials.
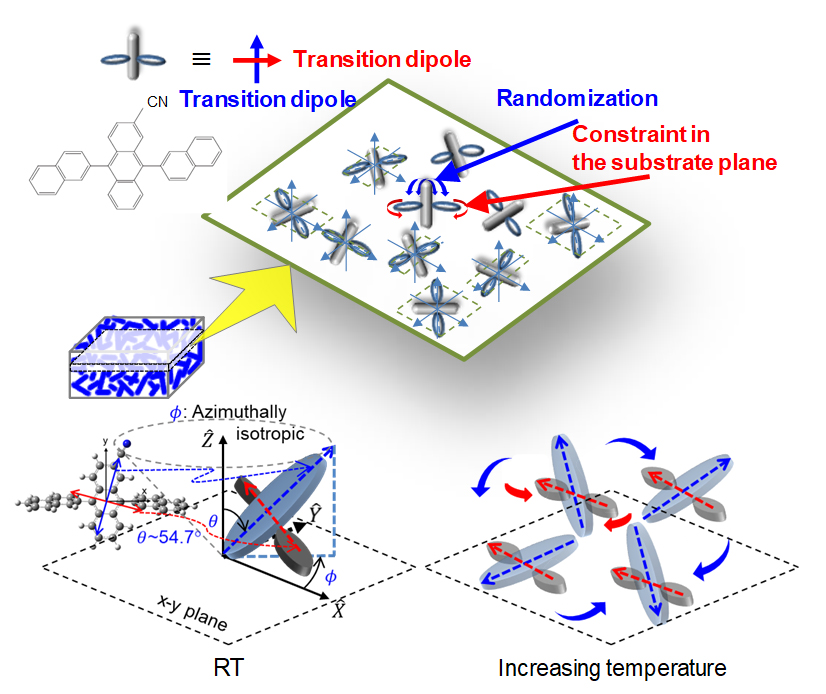
Novel discovery in molecular alignment and orientation of an organic semiconductor molecule while temperature changes.
We observe potential randomization and constraint of molecular alignment and orientation in an organic semiconductor molecule with increasing temperature up to the phase-transition temperature. Variable-angle spectroscopic ellipsometry and second-harmonic generation (SHG) are used to study changes in the molecular alignment in vapor-deposited organic thin films as samples are heated and cooled in a cycle from room temperature to the phase-transition temperature. The films consist of sterically bulky and cross-shaped molecules, 2-cyano-9,10-di(2-naphthyl)anthracene (CADN), and the anisotropy of its two moieties is probed. Anisotropic molecular alignment with respect to the surface normal in as-deposited amorphous films changes with the film thickness, which increases slightly with increasing substrate temperature. Moreover, the long axis of the anthracene moiety changes significantly with respect to the surface normal from the magic angle to isotropic alignment, showing monotonically decreasing anisotropy. Interestingly, the anisotropy of the long axis of the anthracene moiety disappears before the phase-transition temperature. In contrast, the short axis of the anthracene moiety exhibits a notable characteristic change in the temperature dependent alignment during the heating process; although the anisotropy initially decreases, it significantly increases as the temperature approaches the phase transition. At a certain temperature during heating, the film thickness shows a discontinuous jump, similar to a first phase transition, while the anisotropic molecular alignment completely disappears. During the cooling process after the phase transition, however, properties of the films are irreversibly changed, and anisotropic molecular alignment is no longer observed; thus, the samples become completely isotropic.
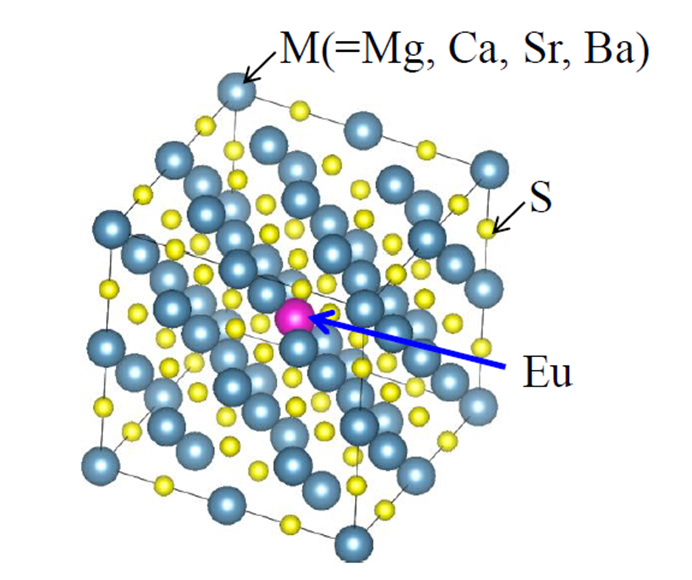
Significantly new approach to analyze photoluminescence properties: Electronic ground state analysis of phosphors
This study theoretically models the electronic states of a series of Eu(II)-doped alkali-earth sulfide phosphors using density functional theory calculations to investigate characterize their photoluminescence properties in terms of quantum efficiency and its thermal decay tendency. We believe that our study makes a significant contribution to the literature because we propose a practical and effective parameter that explains the rankings of quantum yield and its temperature dependence in three of the four phosphor systems, which follow the Dorenbos thermal quenching model. Specifically, this parameter is a ratio of probability amplitudes that reflects the degree to which electrons are localized. Further, we believe that this paper will be of interest to the readership of your journal because our ab initio calculations help to identify the physical reasons underlying the thermal quenching of photoluminescent materials. The parameter presented herein will potentially help other researchers theoretically probe the physics underlying the photoluminescence of phosphors and predict their thermal quenching properties.
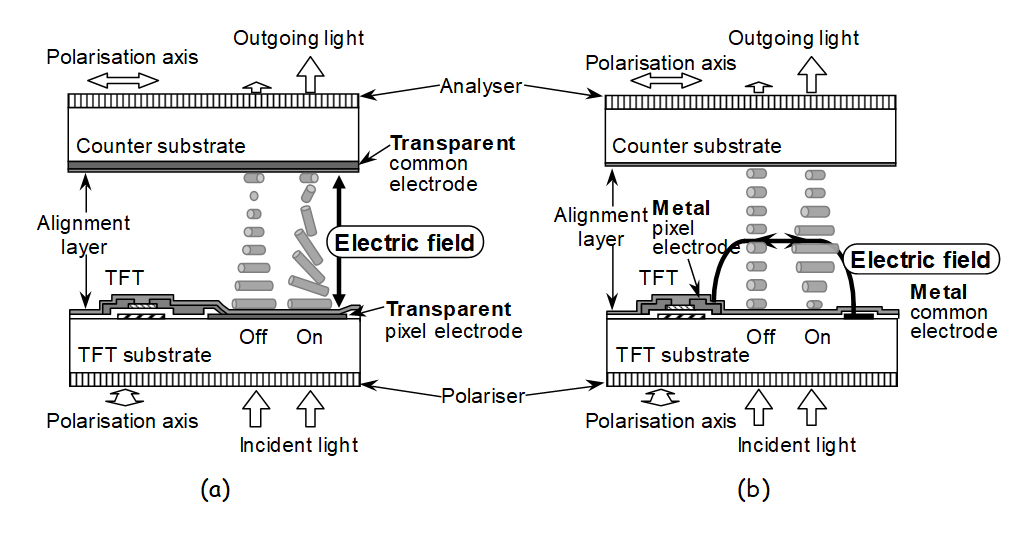
Review of in-plane switching technology as summary of an epoch-making technology in LCDs
We present an overview of in-plane switching (IPS) technologies in terms of historical background, nascent fundamental findings and developments, breakthroughs in addressing display performance issues, and the evolution of materials and device structures. The focus is on how IPS technology has been developed along with changes in the required display characteristics as the target display products continue to evolve. In addition, innovative manufacturing processes, such as photo-alignment technology, have led to the further evolution of IPS technology. Starting with the fundamental frameworks at an early stage, the evolution and development of IPS technology are described as extensively as possible.